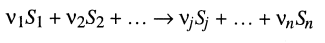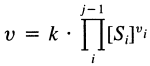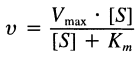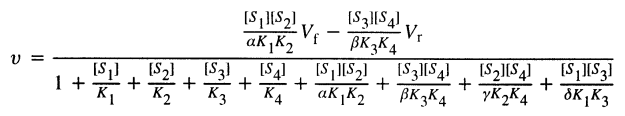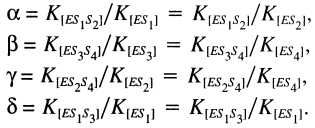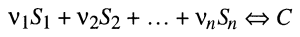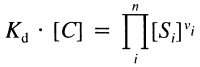E-CELL: software environment for whole-cell simulation
E-CELL: software environment for whole-cell simulation
Masaru Tomita1, Kenta Hashimoto1, Kouichi Takahashi1, Thomas Simon Shimizu1,3, Yuri Matsuzaki1, Fumihiko Miyoshi1, Kanako Saito1, Sakura Tanida1, Katsuyuki Yugi1, J. Craig Venter2 and Clyde A. Hutchison III2
1Laboratory for Bioinformatics, Keio University, 5322 Endo, Fujisawa, 252, Japan and 2The Institute for Genomic Research, 9712 Medical Center Drive, Rockville, MD 20850, USA
Received on May 1, 1998;revised on October 16, 1998; accepted on November 2, 1998
Abstract
Motivation: Genome sequencing projects and further systematic functional analyses of complete gene sets are producing an unprecedented mass of molecular information for a wide range of model organisms. This provides us with a detailed account of the cell with which we may begin to build models for simulating intracellular molecular processes to predict the dynamic behavior of living cells. Previous work in biochemical and genetic simulation has isolated well-characterized pathways for detailed analysis, but methods for building integrative models of the cell that incorporate gene regulation, metabolism and signaling have not been established. We, therefore, were motivated to develop a software environment for building such integrative models based on gene sets, and running simulations to conduct experiments in silico. Results: E-CELL, a modeling and simulation environment for biochemical and genetic processes, has been developed. The E-CELL system allows a user to define functions of proteins, protein-protein interactions, protein-DNA interactions, regulation of gene expression and other features of cellular metabolism, as a set of reaction rules. E-CELL simulates cell behavior by numerically integrating the differential equations described implicitly in these reaction rules. The user can observe, through a computer display, dynamic changes in concentrations of proteins, protein complexes and other chemical compounds in the cell. Using this software, we constructed a model of a hypothetical cell with only 127 genes sufficient for transcription, translation, energy production and phospholipid synthesis. Most of the genes are taken from Mycoplasma genitalium, the organism having the smallest known chromosome, whose complete 580 kb genome sequence was determined at TIGR in 1995. We discuss future applications of the E-CELL system with special respect to genome engineering. Availability: The E-CELL software is available upon request. Supplementary information: The complete list of rules of the developed cell model with kinetic parameters can be obtained via our web site at: http://e-cell.org/. Contact: mt@sfc.keio.ac.jp
The complete genomes of more than 18 microorganisms have been sequenced. The availability of this new information on the gene content of organisms has led to the emergence of a number of heretofore unavailable approaches to biology. Systematic analyses of genes/proteins are now under way in numerous centers around the world, and comprehensive catalogues of protein function are being constructed.
The challenge created by genomics is to understand how all the cellular proteins work collectively as a living system. By attempting to understand the dynamics in living cells, we should be able to predict consequences of changes introduced into the cell and/or its environment, e.g. knocking out a gene or altering available metabolites. Possible consequences of such intervention include cell death, changes in growth rate, and an increase or decrease in the expression of specific genes. The development of sufficiently refined cell models which allow predictions of such behavior would complement the experimental efforts now being made systematically to modify and engineer entire genomes.
In this paper, we present E-CELL, a computer software environment for modeling and simulation of the cell. The E-CELL system is a generic object-oriented environment for simulating molecular processes in user-definable models, equipped with graphical interfaces that allow observation and interaction. E-CELL provides a unified, object-oriented framework for modeling and simulation of the complex interactions among the gene products of completed genomes. Our modeling approach described in this paper attempts to link diverse cellular processes such as gene expression, signaling and metabolism, to construct a cell model for conducting experiments in silico.
Many attempts have been made to simulate molecular processes in both cellular and viral systems. Perhaps the most active area of cellular simulation is the kinetics of biochemical metabolic pathways. Several software packages for quantitative simulation of biochemical metabolic pathways, based on numerical integration of rate equations, have been developed, including GEPASI (Mendes, 1993, 1997), KINSIM (Barshop et al., 1983; Dang and Frieden, 1997), MIST (Ehlde and Zacchi, 1995), METAMODEL (Cornish-Bowden and Hofmeyr, 1991) and SCAMP (Sauro, 1993).
In predicting cell behavior, the simulation of a single or a few interconnected pathways can be useful when the pathway(s) being studied is relatively isolated from other biochemical processes. However, in reality, even the simplest and most well-studied pathways, such as glycolysis, can exhibit complex behavior due to connectivity. Moreover, simulations of metabolic pathways alone cannot account for the longer time-scale effects of processes such as gene regulation, cell division cycle and signal transduction.
Several groups have proposed and analyzed gene regulation and expression models by simulation (Meyers and Friedland, 1984; Koile and Overton, 1989; Karp, 1993; Arita et al., 1994; McAdams and Shapiro, 1995). The cell division cycle (Tyson, 1991; Novak and Tyson, 1995) and signal transduction mechanisms (Bray et al., 1993) have also been active areas of research for biological modeling and simulation. Most of them have utilized qualitative models to deal with the general lack of quantitative data in molecular biology. However, while qualitative models are generally useful when information is incomplete (Kuipers, 1986), they often generate ambiguous results (Kuipers, 1985), the behaviors of which are difficult to predict due to combinatorial explosion (for a review on computer simulations in biology, see Galper et al., 1993).
Previous studies in biochemical and genetic simulations have usually limited their models to focus on only one of the several levels of the time-scale hierarchy in cellular processes. Linking the gaps between the various levels of this hierarchy is an extremely challenging problem that has yet to be adequately addressed. This paper presents a step towards integrative simulation of several levels of cellular processes.
The E-CELL system is, in essence, a rule-based simulation system and is written in C++, an object-oriented programming language. The model consists of three lists, and is loaded at runtime. The substance list defines all objects which make up the cell and the culture medium. The rule list defines all of the reactions which can take place within the cell, and the system list defines spatial and/or functional structure of the cell and its environment. The state of the cell at each time frame is expressed as a list of concentration values of all substances within the cell, along with global values for cell volume, pH and temperature. The simulator engine generates the next state in time by computing all of the functions defined in the reaction rule list. In addition to using the sample models provided with the system, the user can create user-defined models by writing original substance and rule lists. Graphical interfaces are provided to allow observation and interaction throughout the simulation process.
A substance can be a substrate, product or catalyst of a reaction. Typical substances include proteins, protein complexes, DNA (genes), RNA and small molecules. The list of substance concentrations is updated with the new values computed by the simulator engine after each time interval.
In a single time interval, each rule in the rule list is called upon by the simulator engine to compute the change in concentration of each substance. The net change in concentration for each substance is added to the present concentration at the end of each time interval to update the set of state variables, i.e. to generate the next state of the cell. By encapsulating numerical integration methods into object classes, virtually any integration algorithm can be used for simulation of an E-CELL model. Furthermore, E-CELL allows the assignment of any numerical integration algorithm for each compartment of the cell model, facilitating the optimization of the simulation for the user's purpose (e.g. simulation accuracy or speed). Different time intervals ([Delta]t) can also be defined for each spatial or functional compartment and they can be redefined through the control panel at runtime by the user. In the present version, the system defaults to 1 ms for [Delta]t and the user can select between the first-order Euler [error is O([Delta]t2)] or fourth-order Runge-Kutta [O([Delta]t5)] methods for the numerical integration in each compartment. The Euler method is used in compartments with discrete, stochastic reactions such as DNA-protein binding, and the Runge-Kutta method is used for compartments with deterministic reactions defined by continuous rate functions.
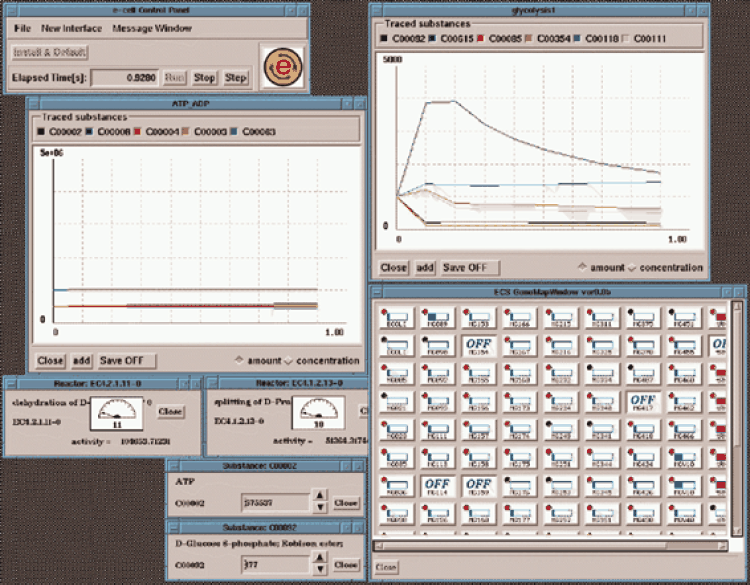
Figure1. A snapshot of user interfaces of the E-CELL system. The tracer window for `glycolysis1' (upper right) shows dynamic changes in quantities of glycolytic metabolites: d-glucose 6-phosphate (C00092), protein histidine (C00615), d-fructose 6-phosphate (C00085), d-fructose 1,6-biphosphate (C00354), d-glyceraldehyde 3-phosphate (C00118) and glycerone phosphate (C00111). The other tracer window (left) shows changes in quantities of ATP (C00002), ADP (C00008), NADH (C00004), NAD+ (C00003) and CTP (C00063). Two reactor windows (lower left) show activities of phosphopyruvate hydratase (EC 4.2.1.11) and fructose-biphosphate aldolase (EC 4.1.2.13). Two substance windows (bottom left) show precise quantities of ATP (C00002) and d-glucose 6-phosphate (C00092). The GeneMapWindow (bottom right) shows current activities (the number of mRNA molecules) of all genes in the cell. Different colors indicate an increase or decrease of activities. Knocked-out genes are marked `OFF'.
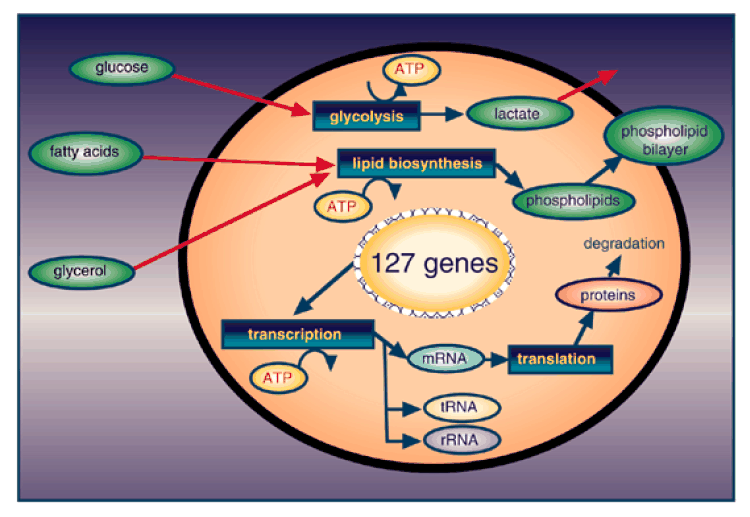
Figure2. Metabolism overview of the model cell. It has pathways for glycolysis and phospholipid biosynthesis, as well as transcription and translation metabolisms.
The simulation of our present whole-cell model runs at ~1/20 of real time on a laptop computer with Pentium-II 200 MHz, and about four times faster on a DEC alpha 21264A 533 MHz with 1 ms integration step and monolithic integration model. A single pathway such as glycolysis runs ~30 times faster under the same conditions.
The E-CELL system provides several graphical interfaces which allow the user to observe the cell's state and manipulate it interactively (Figure 1). The tracer interface is the most important interface which allows the user to select substances or reactions of interest and observe dynamic changes in their quantity or rate, respectively. Since the state of the cell in an E-CELL simulation is defined as the list of all substance quantities, this interface provides the most direct means of observing the cell. Observing dynamical changes in reaction rates is equally important, as the systemic behavior of the cell is characterized by the interaction of a large number of individual reactions. The tracer interface is implemented as a window displaying a two-dimensional plot in which animated line graphs represent changes in the quantity of selected substances or reactions. Each window can display up to six substances simultaneously, and multiple tracers may be invoked to observe all substances of interest. This interface can also produce a `dump file' of traced data for further analysis.
The substance window shows the exact quantity of a selected substance. It also allows the user to alter the quantity at will during the simulation process. The reactor window displays the activity of a selected reaction. The activity of a reaction is defined as the amount of product produced in the reaction per second. The gene map window provides the user with a means of monitoring the expression level of all genes at a glance by graphically displaying the quantity of mRNA transcripts for each gene. The gene map window also allows the user to knock out a selected gene or group of genes by a click of the mouse.
In constructing E-CELL, the primary focus of our interest is to develop a framework for constructing simulatable cell models based on gene sets derived from completed genomes. As a first step, we are constructing a model of a hypothetical, minimal cell, based on the gene set of Mycoplasma genitalium, the self-replicating organism having the smallest known genome, whose complete 580 kb genome sequence was determined in 1995 (Fraser et al., 1995). We have reduced M.genitalium's gene set to accommodate only those genes required for what we have defined, for our purpose here, as a minimal cellular metabolism.
This model cell takes up glucose from the culture medium using a phosphotransferase system, generates ATP by catabolizing glucose to lactate through glycolysis and fermentation, and exports lactate out of the cell. Since enzymes and other proteins are modeled to degrade spontaneously over time, they must be constantly synthesized in order for the cell to sustain `life'. The protein synthesis is implemented by modeling the molecules necessary for transcription and translation, namely RNA polymerase, ribosomal subunits, rRNAs, tRNAs and tRNA ligases. The cell also takes up glycerol and fatty acid, and produces phosphatidyl glycerol for membrane structure using a phospholipid biosynthesis pathway (Figure 2). The model cell is `self-supporting', but not capable of proliferating; the cell does not have pathways for DNA replication or the cell cycle.
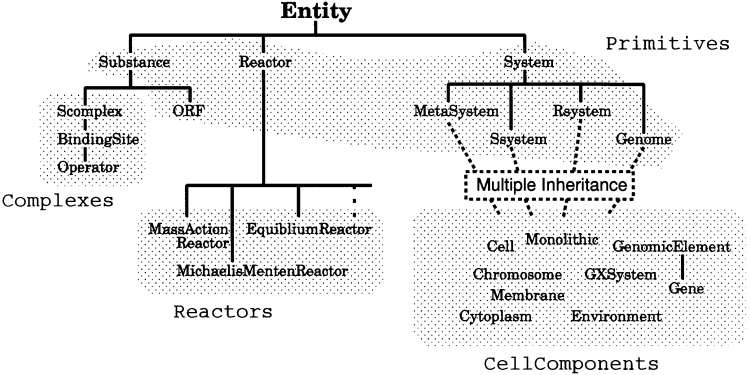
The cell model is basically constructed with three classes of objects: Substances, Genes and reaction rules. The reactions rules are internally represented as Reactor objects. The entire ontology structure of the system is shown in Figure 3.
Figure3. Ontology structure of the E-CELL system. There are three fundamental classes: Substance, Reactor and System. Reactors and CellComponents are the user-definable classes. See our web site for more detailed information.
Substances
All molecular species within the cell are defined as Substances. The same molecule in different states (e.g. phosphorylation) is defined as separate molecular species, and each spatial compartment of the model retains a list of all of the substance objects it may contain.
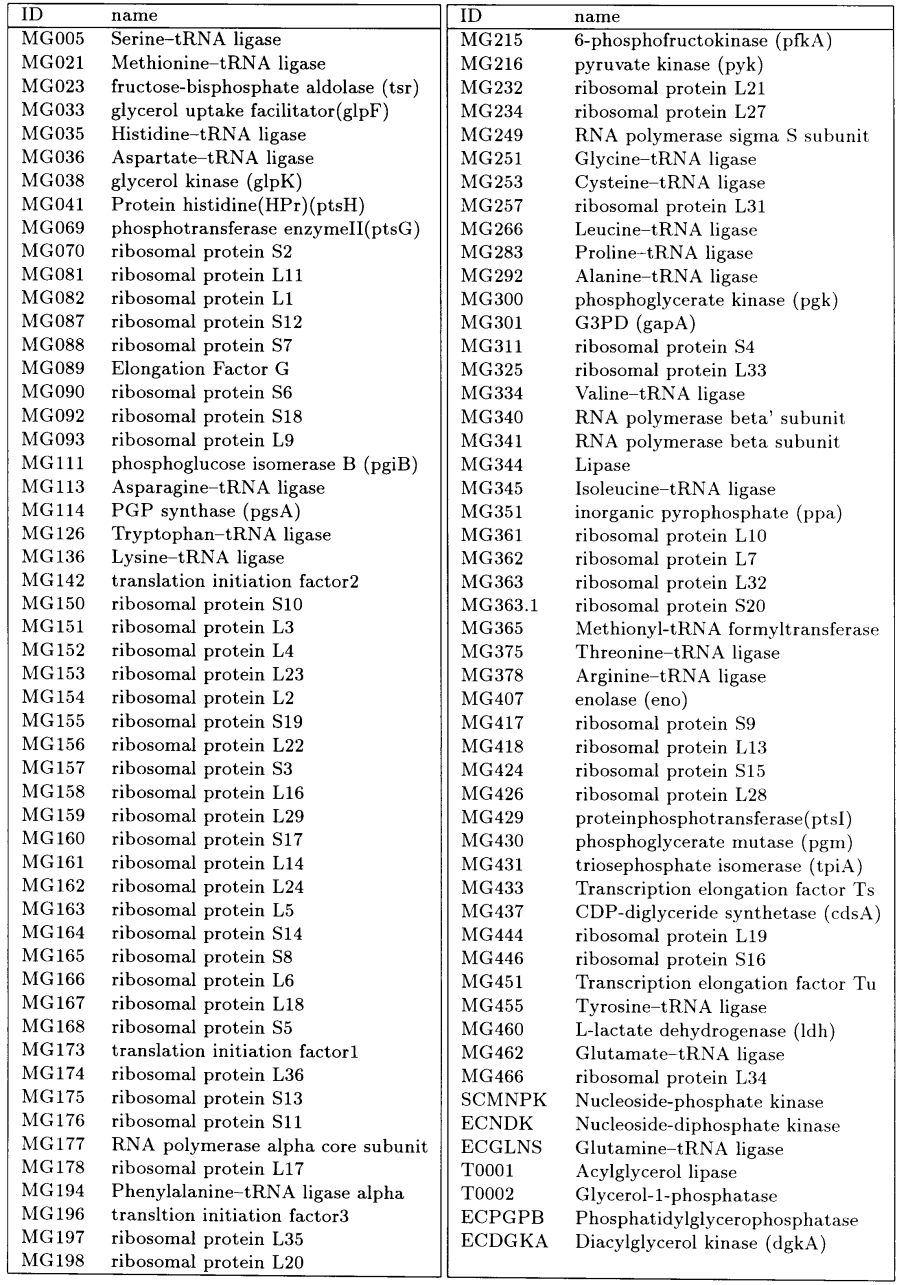
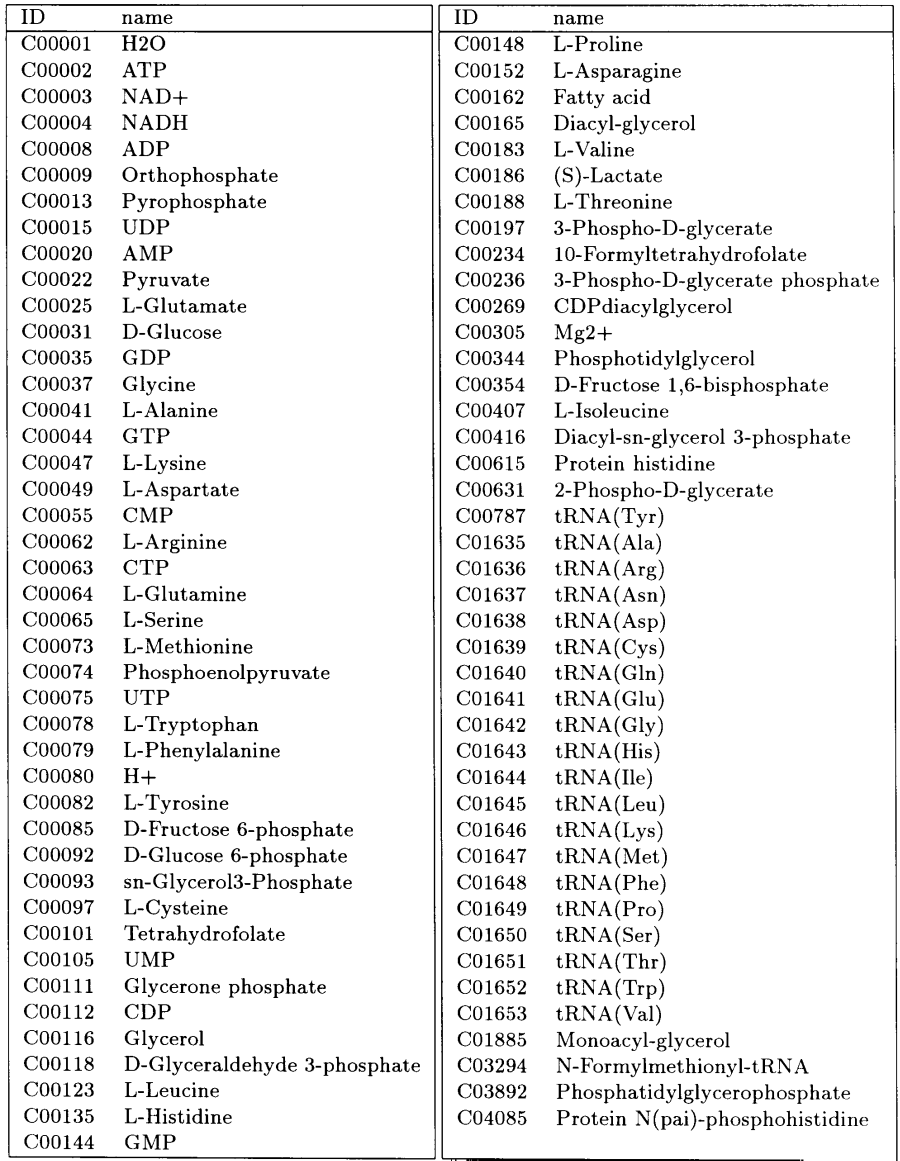
All of the enzymes in our hypothetical model cell are listed in Table 3 and the other small-molecule Substances present in the cell, such as intermediate metabolites, amino acids, nucleotides and cations, are listed in Table 4. Multi-protein complexes, protein-DNA complexes, protein-RNA complexes and other multi-molecule complexes are also defined as Substances, although they are not listed in the table.
Genes
DNA sequences in chromosomes are modeled as a doubly linked list of GenomicElements. The GenomicElement class can have fragments of sequence such as coding sequences, protein binding sites and intergenic spacers. The Gene class is defined as a GenomicElement which has a transcribed sequence.
The genome of the cell consists of 127 genes including 20 tRNA genes and two rRNA genes. Out of the 127 genes, 120 have been identified in the genome of M.genitalium (Table 1 and 2). Four of the seven genes which have not been identified in M.genitalium are for the phospholipid biosynthesis pathway (acylglycerol lipase, glycerol-1-phosphatase, phosphatidylglycerophosphatase and diacylglycerol kinase). The phospholipid biosynthesis pathway of M.genitalium is not well characterized and it is not clear how the functions of these genes are substituted for. Nucleoside-phosphate kinase and nucleoside-diphosphate kinase have also not been identified in M.genitalium, but we have added them to the cell model in order to compensate for the lack of a nucleotide biosynthesis pathway; these enzymes provide a recycling mechanism for degraded DNA/RNA in the model cell, accounting for the lack of nucleotide biosynthesis. The last of the seven E-CELL genes not found in M.genitalium is glutamine-tRNA ligase, whose function is probably substituted for by glutamate-tRNA ligase in M.genitalium, as it is in Gram-positive bacteria (Fraser et al., 1995).
A typical reaction in a metabolic pathway is transformation of one molecular species into another, catalyzed by an enzyme which remains unaltered. For example, the enzyme 6-phosphofructasokinase (EC 2.7.1.11) catalyzes the transformation of d-fructose 6-phosphate (C00085) into d-fructose 1,6-biphosphate (C00354), consuming ATP (C00002) and generating ADP (C00008) and H+ (C00080) (E-CELL Substance IDs shown in parentheses). Schematically, such a reaction can be defined in an E-CELL reaction rule as follows:
Pathways can then be implemented by defining a series of reactions which use the products of another reaction as participating reactants.
The binding reaction of two or more molecules to form a complex can be expressed in a similar way, where the resulting complex would be defined as a separate molecular species. For example, the reaction in which a GTP (C00044) molecule binds to elongation factor Tu (GXtleftu) can be defined as follows:
where `GXtleftu+GTP' is a Substance object representing the complex. Other molecular binding phenomena, such as protein-DNA interaction and ribosome formation from ribosomal proteins, can be modeled in a similar fashion.
Besides quantitative information for each substance, information concerning the location of a substance is often important. We have defined the same molecular species at two different locations as two different objects. For example, the uptake of glycerol (C00116) into the cytoplasm catalyzed by the membrane protein GlycerolUptake PassiveTransport (Egu001) is defined as:
ENVIRONMENT:C00116 -> CYTOPLASM:C00116
[Egu001]
where ENVIRONMENT:C00116 and CYTOPLASM:C00116 represent glycerol in the environment (culture medium) and cytoplasm, respectively.
Using biological knowledgebases for model construction
In order to obtain efficiently the necessary information to implement the pathways in our cell model, we have been utilizing knowledgebases such as EcoCyc (Karp et al., 1996) and KEGG (Kanehisa, 1996). Both of these knowledgebases provide links between information on genes, enzymes and metabolic pathways which proved essential in our effort to construct a model cell.
KEGG was first used to construct the overall structure of the model cell's metabolism based on the gene set of M.genitalium as determined by Fraser et al. (1995). KEGG has a large collection of species-non-specific metabolic pathway diagrams, and provides the utility of highlighting the enzymes which are known/thought to be present in a species of interest. We retrieved diagrams for all of the metabolic pathways which are present in M.genitalium according to KEGG, and manually constructed a single comprehensive network diagram of M.genitalium (not shown).
For our purpose, EcoCyc proved highly useful in obtaining more detailed information about the enzymes and pathways. Although EcoCyc itself does not include kinetic information, its rich references to the literature enabled us to obtain much of the further information we required to build the model.
Transcription and translation
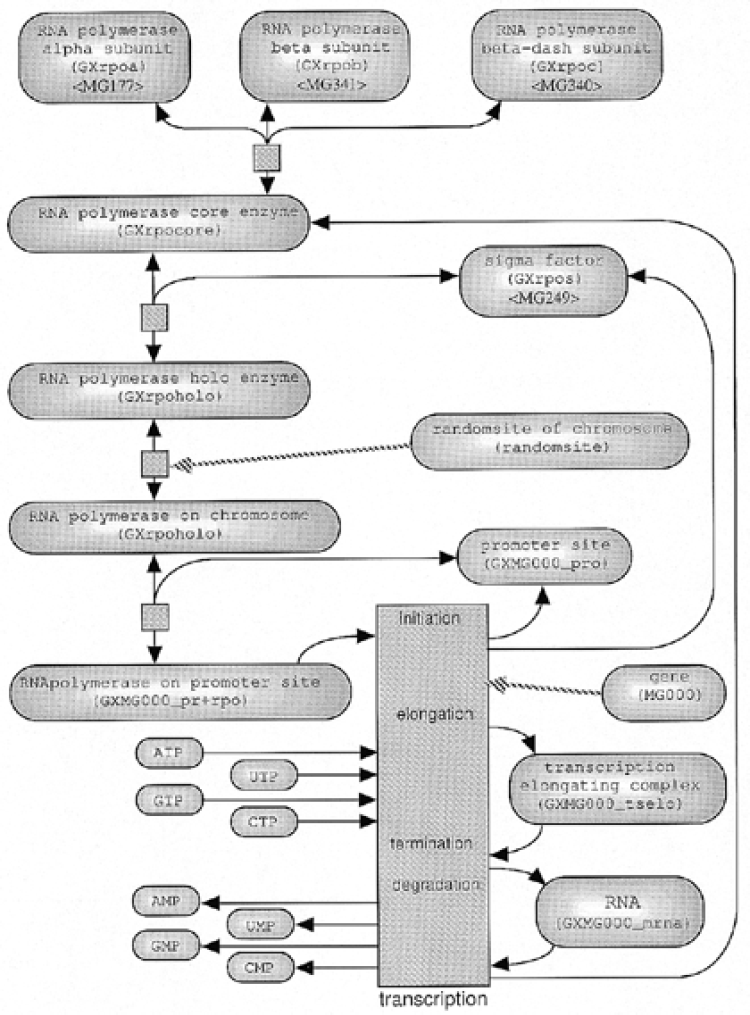
Complex reactions such as transcription and translation are modeled in detail as a series of reactions, part of which is illustrated in Figure 4.
Figure4. The transcription metabolism in the model cell.
Since our present model cell does not need to switch the genes on and off, it does not have any regulatory factors, such as repressors and enhancers. We have therefore not implemented gene regulatory reaction rules, although the software itself allows the user to write rules for sophisticated gene regulatory reactions such as repressor proteins binding to DNA regulatory regions.
Our current model does not utilize actual nucleotide or amino acid sequence information. Although the length of each gene, mRNA and protein is represented, we have made the assumption that each contains equal proportions of nucleotides and amino acids, respectively. In the current cell model, these simplified reaction rules have produced satisfactory results in simulation, and we plan to sustain this level of abstraction until necessary.
Reaction kinetics
Generalizing chemical reactions as:
where Sn is a concentration of the nth substance and [nu]n is a stoichiometric coefficient for the substance, the velocity of each reaction can be expressed as a function of Ss and [nu]s.
Most non-enzymatic reactions are first-order reactions. Their velocities directly depend on concentrations of the substrates and can be expressed as:
where v is the velocity of the reaction and k is the rate constant.
Enzymatic reaction with a substrate and a product can be expressed as the Michaelis-Menten equation:
where [S] is the substrate concentration, Vmax is the maximal velocity of the reaction and Km is the Michaelis constant. One can easily derive equations for reactions involving more than one substrate or product, and incorporate the effects of inhibitor(s) and activator(s) under this Henri-Michaelis-Menten model. For example, the rate equation for a random bi bi reversible enzymatic reaction with an inhibitor and an activator (each product is competitive with each substrate) would be:
where Kn is the dissociation constant for Sn, Vf and Vr are forward and reverse maximal velocity, [alpha], [beta], [gamma] and [delta] are the ratios of dissociation constants of complexes (Kcomplex):
Given a reaction mechanism, such equations can be mechanically derived by hand or with the assistance of computer programs. For more complex enzymatic reactions for which rapid equilibrium assumptions are not inadequate, methods such as the King-Altman method can be used (Segel, 1975).
Some reactions, such as dimer formation and DNA-protein binding, reach equilibrium within a millisecond, which is the default single time unit of the system. For a rapid equilibrium such as:
where C is a complex, the following equation holds at equilibrium:
where Kd is the dissociation constant of the reaction. This equation provides a simple way to compute directly the concentration of each molecular species at equilibrium by only one dissociation constant, i.e. it assumes the binding of more than two Substances to occur simultaneously. However, in reality, the formation of molecular complexes with many components occurs in a stepwise fashion, and in some cellular processes, such as protein signaling, a more detailed representation may be necessary for accurate simulation (Bray et al., 1997). Since we have not implemented any complex signaling pathways in our present cell model, we feel that the use of the simple equation above is justified.
Although some kinetic parameter values can be derived from information available in existing databases, many are unknown. We have assigned values for these parameters by estimations based on available information. Barkai and Leibler (1997) have recently argued that cellular processes are `robust' in many of their properties, in the sense that considerable variation in kinetic parameters often does not affect the behavior of the system as a whole. Many of our simulation results are consistent with their argument; increasing or decreasing a particular parameter by one order of magnitude seldom changes the qualitative behavior of our model cell.
Virtual experiments
The E-CELL interfaces provide a means of conducting `experiments in silico'. For example, we can `starve' the cell by draining glucose from the culture medium. The cell would eventually `die', running out of ATP. If glucose is added back, it may or may not recover, depending on the duration of starvation. We can also `kill' the cell by knocking out an essential gene for, for example, protein synthesis. The cell would become unable to synthesize proteins, and all enzymes would eventually disappear due to spontaneous degradation.
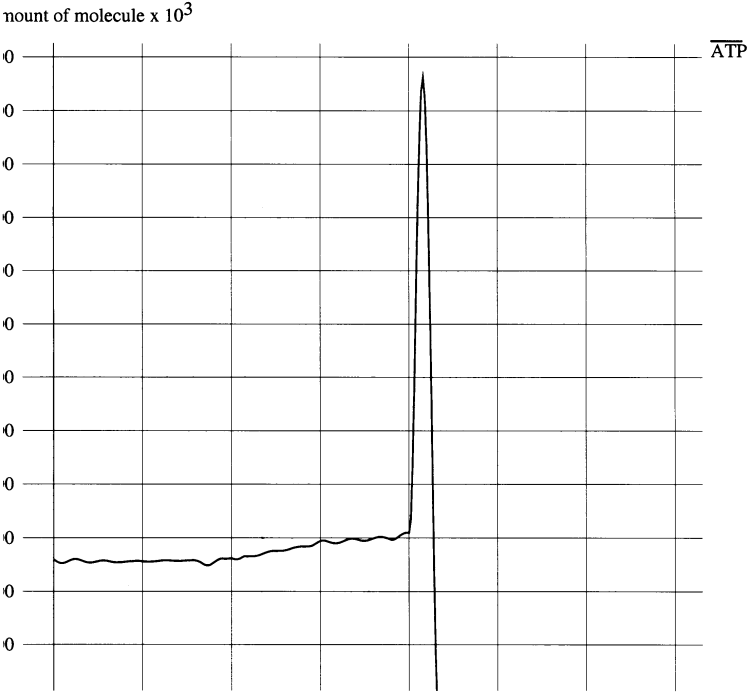
Figure 5 is a trace of the quantity of ATP in the starving cell. Glucose in the culture medium was drained at 20 s. It is interesting that the quantity of ATP temporarily increases at the initiation of starvation. This is explained by the fact that some ATP is consumed in the glycolysis pathway before it produces enough ATP for a net increase. The shortage of glucose to fuel glycolysis arrests the ATP consumption at the beginning of the pathway before the intermediates for ATP production are completely consumed. This results in a temporary increase of net ATP in the cytoplasm. After a short period, however, the quantity of ATP falls sharply.
Figure5. The quantity of ATP increases temporarily and then decreases rapidly when glucose in the culture medium is completely drained at 20 s. The y-axis is the number of ATP molecules (×1000) in the cytoplasm and the x-axis is the elapsed time in seconds.
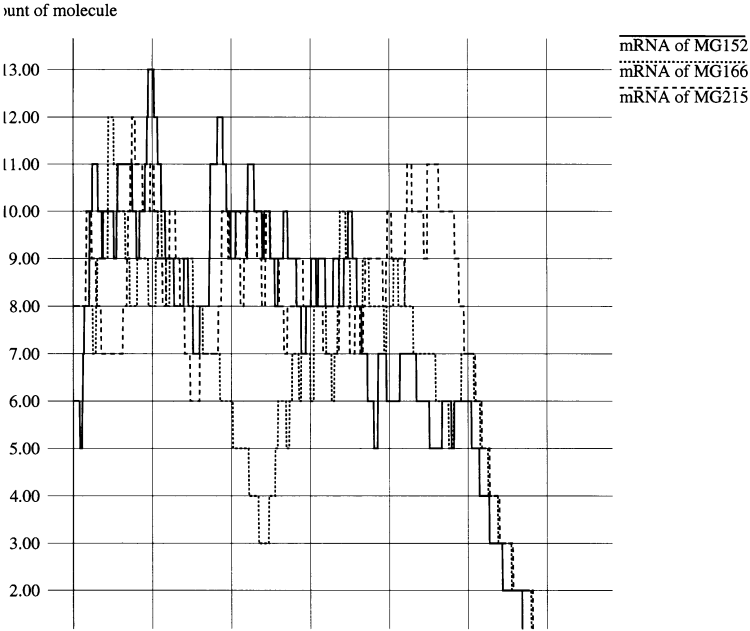
Figure 6 is a trace of the quantity of mRNA, in which the cell was starved at 1000 s. Messenger RNA levels are usually close to steady state due to continuing transcription and degradation. When the cell runs out of ATP after starvation, transcription can no longer continue and mRNAs are rapidly lost by degradation.
Figure6. A trace of mRNA levels before and after starvation of the cell. Before starvation at 1000 s, synthesis by transcription and spontaneous degradation are close to equilibrium. The loss of ATP following starvation causes transcription to stop, and mRNA levels decrease rapidly.
One of our ultimate goals is to model the real cell of M.genitalium, the organism having the smallest known chromosome. Because of the small number of genes (470 proteins, 37 RNAs), M.genitalium is a prime candidate for exhaustive functional (proteome) analysis. Because there are still many genes whose functions are not yet known, it will probably be necessary to hypothesize putative proteins to complement missing metabolic functions, in order for the model cell to work in silico.
Metabolic requirements
The assessment of the metabolic requirements of the cell is an excellent example of a potential application for E-CELL. At present, M.genitalium is grown in a complex medium containing several chemically undefined components including fetal bovine serum, and also extracts of yeast and beef. The problem of designing a chemically defined growth medium could be addressed through a purely empirical approach. However, a more interesting approach is one that is informed by knowledge of the complete genome sequence. By combining knowledge of the metabolic enzymes present in the cell with information concerning protein transporters of metabolites across the cell membrane, it should be possible to evaluate whether a particular defined medium can support growth, by using the E-CELL model. The main difficulty in this approach is that identification of gene function solely on the basis of sequence is uncertain. Comparison of laboratory results with E-CELL predictions should help to overcome this difficulty. Agreement between the model and laboratory growth experiments will be evaluated for a large number of different chemically defined media. Differences between experimental observations and the E-CELL predictions will be used to refine the model. This could lead to the identification of new enzymes or transporters among genes with previously unassigned roles, or to the removal of a questionable role assignment based on a marginal level of sequence similarity.
Gene expression
Another area in which we plan to apply the E-CELL software is in the deciphering of gene regulatory networks. Gene expression patterns of M.genitalium are currently being determined at TIGR under a variety of growth conditions. We expect that these results will suggest specific mechanisms for control of transcript levels which can be modeled by rules in the E-CELL system. We will conduct parallel experiments in the laboratory and in silico with the E-CELL system; given an appropriate model of the cell, we can change initial values of ingredients of the culture medium and observe increases and decreases of mRNA levels. The results of those in silico experiments should be consistent with results of biological and biochemical experiments. The computer model will then be refined as necessary.
Minimal gene set
We expect that the E-CELL system will be useful in defining the minimal set of genes required for a self-replicating cell under a specific set of laboratory conditions. At TIGR, work is under way to identify the genes of M.genitalium which are non-essential, by gene disruption experiments using transposons. If the E-CELL model is sufficiently detailed and accurate, then these gene disruption experiments can be modeled in silico to predict a minimal gene set. The laboratory experiments will lead to the prediction of a reduced gene set which should be a close approximation to the truly minimal Mycoplasma genome. Alternative predictions of a minimal gene set can also be proposed on theoretical grounds, or by deducing a core set of genes conserved between M.genitalium and other microbial genomes. The E-CELL system should be useful in modeling cells based on these alternative proposals for a minimal cellular genome.
We expect that a combination of laboratory experiments and in silico modeling using the E-CELL system will lead to a more reliable prediction of the minimal gene complement for a self-replicating cell than could be obtained by either method alone.
We have constructed a hypothetical cell using the first version of E-CELL, and have developed hundreds of reaction rules for a partial set of metabolic pathways of M.genitalium, including glycolysis, lactate fermentation, glycose uptake, glycerol and fatty acid uptake, phospholipid biosynthesis, gene transcription, protein synthesis, polymerase and ribosome assembly, protein degradation and mRNA degradation.
Our model cell's gene set of 127 genes is much smaller than the `minimal gene set' derived through sequence comparison of the first two sequenced genomes (Fleischman et al., 1995; Fraser et al., 1995) by Musheginan and Koonin (1996). This is not surprising since our model lacks several important features present in all real living cells. The model cell does not proliferate; we are currently modeling cell growth, DNA replication, chromosome segregation and cell division. (The next version of the E-CELL system will have features to support modeling cell division, including dynamic compartment creation/deletion, programmable compartment volume, dynamic reactor/substance creation/deletion, and dynamic DNA sequence representation.)
Furthermore, the present cell model relies on unrealistically favorable environmental conditions. All of the amino acids and nucleotides must exist, and pH and osmolarity must be kept at physiologically stable levels at all times. The model also lacks cell structure proteins, which would be indispensable in any natural environment.
To address these problems, we are currently modeling amino acid and nucleotide biosynthesis pathways. We also plan to model homeostasis of pH and osmolarity, as well as proteins for membrane and cell structure.
An additional point which is worth mentioning is that although simulation is the primary focus of this research, the modeling process has involved much knowledge integration. Although our efforts to gather extensive information on a single organism, M. genitalium, involved much manual methods (e.g. creating diagrams of metabolic networks) and are not, of course, completely automated, we have derived many routine protocols for modeling pathways. We would like to integrate E-CELL's knowledge representation scheme with the schemes of knowledgebases such as EcoCyc and KEGG to facilitate and, where applicable, automate information retrieval, which has proven to be a largely time-consuming part of the modeling process.
The applications of E-CELL, such as genome engineering, have only just begun. The approaches to defining a minimal gene set, described in `User interfaces', are testable in principle. At TIGR a longer term goal of this work is the engineering of the genome to produce living cells with substantially reduced genomes. This will allow us to test proposals for minimal gene sets directly. It will be interesting to compare real cells so created with their computer models. Comparison of the models with the results of laboratory experiments will allow further refinement of the computer models. This, in turn, will lead to a better understanding of the experimental results, and hence a better understanding of the essential requirements of a minimal living cell.
We would like to thank Peter Karp and Doug Brutlag for useful comments on the early draft of this paper. Masanori Arita kindly helped us in reviewing literature on biological simulations. Many of the ideas presented in this paper were inspired by discussions with other E-CELL project members of Tomita Lab. at Keio University, including Junko Shinada, Keiko Miura, Hisako Nakano, Daisuke Kamiyoshikawa, Ryo Matsushima, Akiko Kawase, Naoko Watanabe, Ken Satoyoshi and Yusuke Saito. We also thank for useful comments Scott Peterson and Karen Ketchum of the Institute for Genomic Research, as well as Nobuyoshi Shimizu and his colleagues at the Department of Molecular Biology, Keio University.
This work was supported by the Eizai Research Institute and also in part by a Grant-in-Aid for Scientific Research on Priority Areas `Genome Science' from the Ministry of Education and Science in Japan, as well as a Travelling Fellowship from the Company of Biologists for T.S.S.
Arita ,M., Hagiya,M. and Shiratori,T. (1994) GEISHA SYSTEM: an environment for simulating protein interaction. In Takagi,T. (ed.), Proceedings, Genome Informatics Workshop 1994. Universal Academy Press, Tokyo, pp. 81-89
Barkai ,N. and Leibler,S. (1997) Robustness in simple biochemical networks. Nature, 387, 913-917 MEDLINE Abstract
Barshop ,B.A., Wrenn,R.F. and Frieden,C. (1983) Analysis of numerical methods for computer simulation of kinetic processes: development of KINSIM-a flexible, portable system. Anal. Biochem., 130, 134-145. MEDLINE Abstract
Bray ,D., Bourret,R.B. and Simon,M.I. (1993) Computer simulation of the phosphorylation cascade controlling bacterial chemotaxis. Mol. Biol. Cell, 4, 469-482. MEDLINE Abstract
Cornish-Bowden ,A. and Hofmeyr,J.H. (1991) MetaModel: a program for modeling and control analysis of metabolic pathways on the IBM PC and compatibles. Comput. Applic. Biosci., 7, 89-93.
Dang ,Q. and Frieden,C. (1997) New PC versions of the kinetic-simulation and fitting programs, KINSIM and FITSIM. Trends Biochem. Sci., 22, 317 MEDLINE Abstract
Ehlde ,M. and Zacchi,G. (1995) MIST: a user-friendly metabolic simulator. Comput. Applic. Biosci., 11, 201-207.
Fleischmann ,R.D. et al. (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 269, 496-512. MEDLINE Abstract
Fraser ,C.M. et al. (1995) The minimal gene complement of Mycoplasma genitalium. Science, 270, 397-403. MEDLINE Abstract
Galper ,A.R., Brutlag,D.L. and Millis,D.H. (1993) Knowledge-based simulation of DNA metabolism: prediction of action and envisionment of pathways. In Hunter,L. (ed.), Artificial Intelligence and Molecular Biology. AAAI Press/The MIT Press, CA/MA, pp. 429-436.
Kanehisa ,M. (1996) Toward pathway engineering: a new database of genetic and molecular pathways. Sci. Technol. Jpn, 59, 34-38.
Karp ,P.D. (1993) A qualitative biochemistry and its application to the regulation of the tryptophan operon. In Hunter,L. (ed.), Artificial Intelligence and Molecular Biology. AAAI Press/The MIT Press, CA/MA, pp. 289-324.
Karp ,P.D., Riley,M., Paley,S.M. and Pelligrini-Toole,A. (1996) EcoCyc: encyclopedia of E.coli genes and metabolism. Nucleic Acids Res., 24, 32-40. MEDLINE Abstract
McAdams ,H.H. and Shapiro,L. (1995) Circuit simulation of genetic networks. Science, 269, 650-656. MEDLINE Abstract
Mendes ,P. (1993) GEPASI: a software package for modeling the dynamics, steady states and control of biochemical and other systems. Comput. Applic. Biosci., 9, 563-571.
Mendes ,P. (1997) Biochemistry by numbers: simulation of biochemical pathways with Gepasi 3. Trends Biochem. Sci., 22, 361-363. MEDLINE Abstract
Meyers ,S. and Friedland,P. (1984) Knowledge-based simulation of genetic regulation in bacteriophage lambda. Nucleic Acids Res., 12, 1-9. MEDLINE Abstract
Mushegian ,A.R. and Koonin,E.V. (1996) A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl Acad. Sci. USA, 93, 10268-10273. MEDLINE Abstract
Novak ,B. and Tyson,J.J. (1995) Quantitative analysis of a molecular model of mitotic control in fission yeast. J. Theor. Biol., 173, 283-305.
Sauro ,H.M. (1993) SCAMP: a general-purpose simulator and metabolic control analysis program. Comput. Applic. Biosci., 9, 441-450.
Segel ,I.H. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. John Wiley & Sons, New York.
Tyson ,J.J. (1991) Modeling the cell division cycle: cdc2 and cyclin interactions. Proc. Natl Acad. Sci. USA, 88, 7328-7332. MEDLINE Abstract